【文献翻译】人体端粒酶在稳态和疾病中的调控 |
您所在的位置:网站首页 › human ruins翻译 › 【文献翻译】人体端粒酶在稳态和疾病中的调控 |
【文献翻译】人体端粒酶在稳态和疾病中的调控
|
大家好,今天给大家翻译2020年发表在《MUlECULAR CEll Biology》的综述:Regulation of human telomerase in homeostasis and disease。 摘要端粒酶是一种核糖核蛋白复合体,其催化核心包括端粒酶逆转录酶(TERT)和非编码人端粒酶RNA(HTR),端粒酶RNA是将端粒重复序列添加到染色体末端的模板。端粒酶在人类中的表达仅限于某些类型的细胞,在正常情况下端粒酶水平受到严格控制。在绝大多数人类癌症中都发现端粒酶水平升高,我们最近开始了解癌细胞增加端粒酶活性的机制。相反,降低端粒酶功能的端粒酶相关基因的胚系突变会导致一系列遗传疾病,包括先天性角化不良、特发性肺纤维化和骨髓衰竭。在这篇综述中,我们讨论了人类TERT的转录调控,hTR的加工,端粒酶复合体的组装,端粒酶的细胞定位及其在端粒中的募集,以及端粒酶活性的调节。我们还讨论了端粒酶生物发生的每个步骤与疾病的相关性。 介绍端粒包含位于染色体末端的重复序列,可维持线性染色体的完整性。这些染色体末端必须与其他类型的线性 DNA 区分开来,例如注定需要修复的断裂 DNA 末端。端粒序列被一组蛋白质特异性结合,这些蛋白质共同构成了 Shelterin复合物(图 1a)。  图 1 |端粒酶与端粒处的 shelterin 相互作用。a.端粒酶是一种核糖核蛋白复合物,包含脚手架非编码人端粒酶 RNA (hTR)、端粒酶逆转录酶 (TERT) 和相关辅助因子。 TERT 通过位于假结/模板 (PK/T) 结构域和 hTR 保守区域 4/5 (CR4/5) 结构域的端粒酶 RNA 结合结构域(未显示)与 hTR 结合。 图 1 |端粒酶与端粒处的 shelterin 相互作用。a.端粒酶是一种核糖核蛋白复合物,包含脚手架非编码人端粒酶 RNA (hTR)、端粒酶逆转录酶 (TERT) 和相关辅助因子。 TERT 通过位于假结/模板 (PK/T) 结构域和 hTR 保守区域 4/5 (CR4/5) 结构域的端粒酶 RNA 结合结构域(未显示)与 hTR 结合。包含肌动蛋白、NOP10 和 NHP2 的肌动蛋白复合物与 hTR 的 H/ACA 结构域结合。 H/ACA 结构域包括一个单链铰链 (H-box),后跟一个茎环和 ACA 序列。 hTR 的 H/ACA 结构域还包含一个保守的四核苷酸基序,称为 CAB 盒,它结合蛋白质端粒酶 Cajal 体蛋白 1 (TCAB1)。 hTR 的模板区域与端粒 3' 端链结合。端粒重复由蛋白质复合物 shelterin 结合。两种双链 DNA 结合蛋白,即端粒重复结合因子 1 (TRF1) 和 TRF2,直接与端粒 DNA 结合。 TRF2 与 RAP1(也称为 TRF2 相互作用端粒蛋白 1)相互作用,TRF1 和 TRF2 都与 TRF1 相互作用核因子 2 (TIN2) 相互作用,后者也结合 TPP1。 TPP1 还结合端粒蛋白 1 (POT1) 的单链 DNA 结合蛋白保护,并通过 TERT 的 N 末端结构域 (TEN) 将端粒酶募集到端粒。 b.端粒酶示意图显示了 TERT 结构域与 hTR 和端粒酶辅助因子的关系,改编自人类端粒酶的低温电子显微镜结构。端粒酶采用灵活的 RNA 束缚双叶结构。 hTR 的 H/ACA 结构域由两组肌张力障碍复合物(肌张力障碍蛋白、NHP2、NOP10 和 GAR1)和 TCAB1 结合,包含一个叶。第二叶包含催化核心,其中 hTR 和 TERT 环绕端粒底物。这两个叶由 hTR 的 CR4/5 结构域连接。 CTE,C端延伸; RT,逆转录酶; TRBD,端粒酶 RNA 结合结构域。 Shelterin复合物通过阻止对游离DNA末端产生的 DNA 修复反应来解决末端保护问题。这些反应包括通过非同源末端连接、替代性非同源末端连接或同源重组激活 DNA 损伤反应和修复。为了发挥这些功能,Shelterin的蛋白质亚基募集了许多其他蛋白质辅助因子,并且近年来,基于质谱的方法扩大了Shelterin相关蛋白的范围。染色体末端的第二个关键问题是端粒缩短,这是因为在 5'–3' 方向合成 DNA 的 DNA 聚合酶不完全复制滞后链,从而导致端粒序列随时间逐渐缩短。这个末端复制问题通过端粒酶的进化得到解决,端粒酶是一种核糖核蛋白 (RNP) 复合物,其催化核心由端粒酶逆转录酶 (TERT) 和非编码 RNA、端粒酶RNA成分 (TERC;本文称为人类端粒酶 RNA (hTR)),它作为将端粒重复序列添加到染色体末端的模板。 在单细胞真核生物中,端粒酶组成型表达以实现长期细胞分裂和活力。在后生动物中,尤其是在人类中,端粒酶的表达变得仅限于某些细胞类型或细胞状态。人类端粒酶表达在种系和祖细胞区室中最高,但在许多分化的细胞类型中受到限制。这种受限模式的进化可能部分是为了降低患癌症的风险,因为几乎所有人类肿瘤都显示端粒酶表达增加。降低端粒酶功能的端粒酶通路基因种系突变导致干细胞衰竭疾病先天性角化不良、再生障碍性贫血和特发性肺纤维化。这些遗传学发现表明细胞必须保持端粒酶的最佳水平,该水平足以支持组织稳态但又低到足以降低患癌症的风险。除了严格控制酶水平外,端粒酶还在多个额外步骤中受到调节。 在这篇综述中,我们讨论了有关人类端粒酶复合物如何在正常祖细胞和癌细胞中受到调节以及这些过程如何在疾病中发生改变的最新信息。TERT基因的转录是调节端粒酶表达的关键,并决定哪些细胞类型可以表达端粒酶。需要对 hTR 的 3' 端进行加工才能形成成熟的hTR分子,并且会受到决定稳态端粒酶水平的定量调节。端粒酶是一种大的多亚基RNP,其中包括几种在端粒酶功能中具有不同作用的蛋白质成分。这种复合物的组装需要许多额外蛋白质的活性和发育中的复合物通过细胞核的不同亚区的运动。端粒酶向端粒的募集受特定蛋白质-蛋白质相互作用的控制,并且该酶在端粒结合后受到进一步调节。在癌症发展过程中,TERT 通过多种机制上调,以支持与肿瘤发生相关的增强细胞增殖。端粒维护受损是一系列以细胞耗竭和终末期器官功能障碍为特征的组织表型的基础。对这些疾病状态的研究可以直接研究人体组织中端粒酶功能的生化缺陷。 端粒酶转录和成熟端粒酶活性的关键调节是通过 TERT 转录的控制发生的,它决定了 TERT 水平并决定了表达端粒酶的细胞类型。 hTR 的调节是通过其 3' 端的转录后成熟过程进行的,该过程设定了成熟 hTR 的水平,进而设定了成熟端粒酶复合物的水平(图 2)。 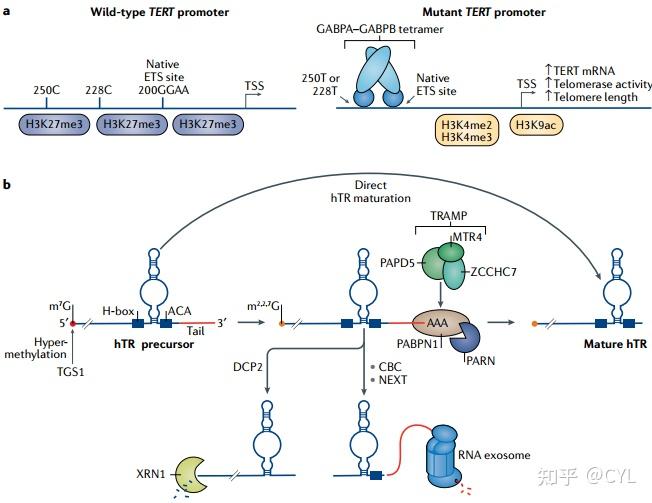 图 2 |端粒酶的 TERT-hTR 催化核心的调节。a.端粒酶逆转录酶 (TERT) 转录的调节和突变在 TERT 启动子中的作用。 TERT 基因的野生型启动子通常被组蛋白 H3 Lys27 (H3K27me3) 修饰的基因抑制三甲基化沉默。野生型启动子包含 ETS 转录因子家族的天然结合位点(带有基序 GGAA). 图 2 |端粒酶的 TERT-hTR 催化核心的调节。a.端粒酶逆转录酶 (TERT) 转录的调节和突变在 TERT 启动子中的作用。 TERT 基因的野生型启动子通常被组蛋白 H3 Lys27 (H3K27me3) 修饰的基因抑制三甲基化沉默。野生型启动子包含 ETS 转录因子家族的天然结合位点(带有基序 GGAA).在不同的癌症类型中,C→T 转换发生在翻译起始密码子上游 124bp 和 146bp 处,根据它们的基因组坐标被称为 C228T 和 C250T。这些突变是相互排斥的,并在远离原生 ETS 位点的三到五个螺旋圈(未显示)处产生额外的 ETS 结合位点,这对于募集 ETS 异四聚体 GABPA–GABPB 是最佳的。突变的 TERT 等位基因通常与 RNA 聚合酶 II 相关,并以基因激活 H3K4me2、H3K4me3 和乙酰化组蛋白 H3 Lys9 (H3K9ac) 修饰为标志。突变的 TERT 等位基因导致 TERT 转录上调和端粒酶活性增加。 TSS,转录起始位点。 人类端粒酶 RNA (hTR) 生物发生的调节。 hTR 被转录为具有 5' 甲基鸟苷帽 (m7 G) 的前体分子。 m7 G 帽被三甲基鸟苷合酶 (TGS1) 进一步甲基化为 m2,2,7G。大多数 hTR 转录本都有一个短的(图 3 |细胞内端粒酶的组装和运输。端粒酶逆转录酶 (TERT) 在分子伴侣热休克蛋白90 (HSP90) 的帮助下折叠,而人端粒酶 RNA (hTR) 与肌张力障碍复合物共转录结合,包括肌张力障碍、NOP10、NHP2和相关因子 NAF1。 辅助因子 pontin 和 reptin 促进 hTR 与肌张力障碍复合物和 TERT 的组装。端粒酶通过端粒酶 Cajal 体蛋白 1 (TCAB1) 定位于 Cajal 体,该蛋白由伴侣蛋白 TRiC 复合物折叠。在没有 TCAB1 的情况下,端粒酶错误定位到核仁(未显示)并且不会被招募到端粒。在 S 期,TERT 的 N 末端结构域 (TEN) 和 TPP1 的寡糖/寡核苷酸结合 (OB) 折叠结构域上的 TEL补丁(未显示)相互作用,将端粒酶募集到端粒。 单分子活细胞成像显示端粒酶在Cajal小体中的停留时间延长。端粒酶在Cajal小体中定位的目的尚不清楚,因为尽管Cajal小体被破坏,但在Colin基因敲除的癌细胞中端粒仍然有效地保持着。此外,在某些小鼠细胞中,端粒酶不定位于Cajal小体。然而,在低水平端粒酶表达的细胞环境中,或者在某些尚未被研究的细胞类型中,Cajal小体仍然可能在调节端粒酶方面发挥重要作用。由于端粒酶研究中的技术挑战,其亚核定位仅在少数细胞类型中被研究。 招募端粒并与Shelterin结合端粒酶向端粒的募集是由 TERT 和 shelterin 蛋白 TPP1 之间的相互作用介导的(图 1a)。TPP1通过TRF1相互作用核因子 2 (TIN2) 与其余的 shelterin 复合物相连,TIN2 也与双链 DNA 结合蛋白端粒重复结合因子 1 (TRF1) 和 TRF2形成接触。TPP1还与端粒蛋白 1 (POT1) 的保护有关,它直接结合单链端粒悬突。TPP1 或 TIN2 的缺失会减少端粒酶向端粒的募集。TERT和TPP1之间的特异性相互作用由 TERT 的十个结构域和 TPP1 寡糖/寡核苷酸结合折叠结构域表面的氨基酸补丁介导,称为 TEL 补丁(图 3)。在TPP1的这个区域发现突变会导致端粒缩短和严重的先天性角化不良,这突显了 TEL 补丁的重要性。单个端粒酶分子的活细胞成像显示端粒酶 RNP 扩散通过细胞核并与端粒形成瞬时和长期相互作用。人类胚胎干细胞中TEL补丁突变导致TERT-TPP1结合丧失,导致端粒缩短和细胞活力丧失,从而强调了端粒酶募集对端粒维持的重要性。有趣的是,在这种情况下,TERT 与 TPP1 的连接不能完全挽救端粒缩短,这表明人类 TPP1 的TEL 补丁在端粒酶激活中具有额外的作用。 TIN2 对于维持端粒长度也是必不可少的;如果没有,TPP1和 POT1无法定位到端粒。由于这种桥接功能,需要 TIN2才能将端粒酶募集到端粒。反映其在端粒长度维持中的重要性,TIN2 在患有短端粒综合征的个体中发生突变,突变聚集在靠近蛋白质 C 末端区域的外显子 6 中。虽然这些突变不影响 TIN2 定位或端粒酶活性,但有一些证据表明它们会损害端粒酶向人类细胞端粒的募集。然而,致病 TIN2-K280E 突变的小鼠模型通过端粒酶独立机制表现出端粒缩短。需要更多的研究来准确阐明这些 TIN2 突变如何导致端粒缩短。端粒蛋白也可以抑制端粒上的端粒酶活性。 CST复合物包含 CTC1、STN1 和 TEN1,是一种保守的三聚体复合物,可结合端粒 3' 突出端(图 4)。 人类CST复合物通过刺激DNA聚合酶 α-primase来协助端粒处的 DNA 复制。此外,CST通过限制端粒酶进入端粒并抑制 TPP1–POT1对端粒酶的刺激来限制端粒延伸(见下文)。因此,CST复合物可促进从端粒酶介导的端粒延长转变为端粒填充合成。 端粒酶向端粒的募集以细胞周期依赖性方式进行调节。在出芽酵母和裂殖酵母中,端粒酶与端粒的相互作用仅限于 S 期。同样,在人类细胞中,端粒酶和端粒的共定位主要发生在 S 期(图 3 和 4)。  图 4 |端粒端粒酶活性的调节。 图 4 |端粒端粒酶活性的调节。a. |募集:端粒酶在 S 期通过 shelterin 复合亚基 TPP1 的寡糖/寡核苷酸结合 (OB) 折叠结构域与端粒酶逆转录酶 (TERT) 的 N 末端结构域 (TEN) 之间的接触被募集到端粒。端粒酶活性受端粒酶 Cajal 体蛋白 1 (TCAB1) 调节,它在称为 CAB 盒的保守四核苷酸序列上与人端粒酶 RNA (hTR) 的 H/ACA 结构域结合。 TCAB1 结合导致 hTR 保守区域 4/5 (CR4/5) 结构域的 P6.1 和 P6b 区域发生构象变化,从而增强端粒酶活性。 hTR 中的 TCAB1 相互作用位点标记为红色。 b.延伸:端粒酶延伸端粒的富含 G 链(橙色)。端粒酶的持续合成能力——端粒酶在一次引物结合事件中可以添加的端粒重复序列的数量——由 shelterin 成分 TPP1 和端粒蛋白 1 (POT1) 的保护作用增强。 TPP1–POT1 通过促进端粒酶沿端粒底物的易位和/或通过防止端粒酶与端粒引物的解离来促进端粒酶的持续合成能力。 c |终止和填充:在晚期 S 期/G2 期,CST 复合物(包括 CTC1、TEN1 和 STN1)结合端粒新合成的 G(-rich) 链并阻止端粒酶接近它。 CST 还会削弱端粒酶与 TPP1–POT1 的相互作用并促进 DNA 聚合酶 α-引物酶的活性,后者合成填充端粒 C 链所必需的 RNA 引物(红线)。 端粒酶的细胞周期调节组装可以解释端粒酶募集到 S 期的一些限制。沿着这些思路,TERT和 pontin之间的关联在细胞周期期间发生变化,并在S期达到峰值。有趣的是,端粒酶复合物与 TCAB1 的组装在细胞周期中发生变化。TCAB1-hTR关联在有丝分裂细胞中丢失,表明端粒酶的某些元素以细胞周期调节的方式组装。 这一观察结果符合先前确定的 TCAB1在端粒酶募集到端粒中的重要性,并表明 TCAB1与端粒酶结合的调节解释了一些细胞周期依赖性募集。因此,端粒酶向端粒的募集需要 TERT 和Shelterin蛋白 TPP1之间的相互作用,并且主要发生在S期;然而,控制这种招募时间和监管的确切机制仍然未知。 端粒酶活性的调节端粒酶活性是否可以通过其蛋白质辅助因子进行调节一直是一个活跃的研究领域。TCAB1 最初被定性为一种端粒酶蛋白,是端粒酶运输到 Cajal体和维持端粒长度所需的蛋白.在使用基因组编辑删除TCAB1 的人类和小鼠细胞中,端粒酶催化活性受损。这些细胞中端粒酶活性的降低是由 hTR CR4/5 结构域内关键 RNA 螺旋的部分展开引起的,这反过来又削弱了它与 TERT的结合。这些发现表明,TCAB1 的结合通过增强 RNA 折叠和促进 TERT 和 hTR 的正确结合来促进端粒酶的催化活性状态(图 4a)。 一旦被招募到端粒,端粒酶必须被进一步激活以有效地添加端粒重复序列。端粒端粒酶的调节是通过 shelterin 成分的活性进行的。由于 TPP1-POT1 通过 POT1与单链 DNA 结合,最初的预测是端粒酶和 TPP1-POT1将竞争端粒结合,并且 TPP1-POT1 将通过隔离 3' 突出端来抑制端粒酶活性。为支持这一假设,过表达不能结合单链 DNA 的 POT1 突变体可通过增强端粒处的端粒酶活性导致端粒快速延长.与这一发现相反,在体外端粒酶中加入重组TPP1和POT1,端粒酶的持续合成能力增加了两到三倍(定义为在单一端粒酶-引物结合事件后增加的端粒重复数)。进一步的研究表明,TPP1–POT1 通过防止端粒酶与端粒DNA的解离或通过增加每次重复添加后端粒酶易位的效率来改善端粒酶的体外持续合成能力(图 4b)。端粒酶持续合成能力的刺激由 TPP1的TEL补丁介导,它也在端粒酶募集中发挥作用. 结合重组 TPP1–POT1,添加外源性 TIN2 也可以刺激细胞中的端粒酶持续合成能力。尽管缺乏对端粒酶持续合成能力的体内研究,但从理论上讲,端粒酶的这种激活机制允许端粒酶在体外仅通过少数引物结合事件合成长链端粒 DNA。需要进一步研究以更好地理解 TPP1–POT1 在增强和抑制端粒酶功能方面的双重作用。 端粒酶上调几乎普遍存在于人类癌症中端粒酶的上调或再激活发生在超过 90%的人类肿瘤中。虽然端粒酶似乎在大多数增殖的祖细胞中表达,但端粒酶的量不足以在衰老和体内平衡期间维持端粒。端粒酶水平不足可以解释端粒在衰老过程中在人体组织中缩短的普遍现象。如上所述,在实验室中培养的许多类型的原代人类细胞完全缺乏端粒酶,并显示出随着细胞分裂进行性端粒缩短。经过几个月的长期滞后期,人类成纤维细胞停止分裂并进入复制衰老状态。我们现在了解到,复制性衰老是由一部分非常短的端粒引起的,这些端粒失去了保护染色体末端的能力,被认为是受损的 DNA,这会诱导 DNA 损伤反应,包括p53和RB肿瘤抑制通路的激活。p53和RB 的失活延长了端粒极短的细胞的增殖,但最终导致端粒危机:基因组不稳定和细胞死亡的时期。虽然端粒危机会损害细胞分裂,但它也可以通过诱导非相互易位、非整倍体和拷贝数变化来驱动恶性转化。端粒危机期间诱导的细胞死亡是由自噬驱动的。在成纤维细胞中,TERT 的过表达重组端粒酶,防止细胞衰老和危机,并使细胞永生。人类肿瘤中端粒酶的上调具有赋予细胞永生化的相同功能,从而促进肿瘤侵袭和转移。 端粒酶通过多种机制在癌症中上调,包括TERT 和hTR 编码基因TERC的基因扩增。癌症增加TERT转录的最佳理解机制是通过获得TERT启动子中的非编码突变(图 2a)。TERT 启动子突变代表了几种肿瘤类型中最常见的突变,包括胶质母细胞瘤、黑色素瘤、膀胱癌和肝细胞癌。此外,它们是人类癌症中最常见的非编码突变。TERT 启动子突变激活TERT转录,导致尿路上皮癌细胞系中mRNA水平升高、端粒酶活性升高和端粒长度增加。两个常见的C→T转换突变位于近端启动子,翻译起始密码子上游124bp和146bp。根据它们的hg19 基因组坐标(chr5, 1,295,228 C>T 和 chr5, 1,295,25 C>T),这些突变被称为 C228T 和 C250T。C228T 和 C250T 突变是杂合的,相互排斥并为 ETS家族转录因子创建从头结合位点(图 2a)。 突变的TERT启动子专门募集 ETS 家族转录因子GABPA及其辅助因子GABPB。这两个转录因子形成一个异源四聚体,它与TERT 启动子的基序以及突变产生的新基序结合。在携带TERT启动子突变的癌细胞中,TERT mRNA 特异性地来源于突变的等位基因。与这一发现一致,野生型等位基因仍然标记为三甲基化组蛋白 H3 Lys27 (H3K27me3),这是一种基因抑制组蛋白修饰,而突变等位基因标记为基因活性组蛋白修饰 H3K4me2、H3K4me3和乙酰化组蛋白 H3 Lys9 (H3K9ac)(图 2a)。因此,TERT 启动子中的突变可能会阻止启动子的正常沉默。在表达端粒酶的人胚胎干细胞中产生这些 TERT 启动子突变导致 TERT mRNA 水平适度增加。然而,在分化成成纤维细胞后,突变细胞保留了端粒酶表达,而同基因对照细胞系使 TERT沉默。 TERT启动子突变也可能改变 TERT 基因座的远程接触以促进转录激活。 TERT 在癌症中上调的频率表明它对肿瘤发生至关重要。基于TERT启动子突变仅限于自我更新率通常较低的组织(如中枢神经系统、肝脏和黑色素细胞)的癌症,以及它们不存在于增殖性更强的组织(如结肠和血液)的癌症中,有人提出,具有低基础端粒酶活性的组织需要在肿瘤发生早期激活端粒酶突变。支持这一论点的是,TERT启动子突变发生在癌症的早期前驱病变中,例如肝细胞癌、膀胱癌、皮肤黑色素瘤、甲状腺癌、鳞状细胞癌、基底细胞癌和少突胶质细胞瘤。 除了选择TERT启动子突变外,癌症还通过多种机制上调TERT转录。在某些肝细胞癌病例中,乙型肝炎病毒 (HBV) 可以将其基因组整合到紧靠TERT启动子的位置,从而将病毒增强子元件放置在启动子附近而不破坏编码序列。神经母细胞瘤中的染色体重排将 TERT 基因座定位在强增强子元件附近,从而上调TERT转录。此外,在一组近7000个肿瘤样本中,4%的人有TERT基因扩增,最常见的是卵巢癌和肺腺癌样本。重要的是,胶质母细胞瘤中的 TERT 启动子突变和 TERT 重排均与 ATRX 或 DAXX 中的突变相互排斥,ATRX 或 DAXX 是组蛋白伴侣,通常在癌细胞中失活,采用不依赖于端粒酶的端粒维持机制,称为端粒替代延长 (ALT) 。ALT 基于同源重组并使用断裂诱导复制来合成端粒 DNA的轨迹。癌细胞中的 ALT 导致长而异质的端粒和缺乏端粒酶表达 。ALT 是胶质母细胞瘤、神经内分泌肿瘤和某些肉瘤中端粒维持的常见机制。 癌症中 hTR 的上调研究较少,但一些证据表明,与 TERT 一样,hTR在人类肿瘤中被转录激活。已在多种癌症类型中检测到 hTR 编码基因TERC的拷贝数增加,包括宫颈癌、卵巢癌和肺癌。最后,尽管据估计超过 90% 的癌症会上调端粒酶活性,但大多数端粒酶阳性肿瘤缺乏 TERT 启动子突变。一项对超过 18,000 个肿瘤和匹配的正常组织样本的测序数据的研究发现,在表达 TERT 的肿瘤中,32% 携带 TERT 启动子突变、基因扩增或染色体重排 。这些观察表明,具有可检测的端粒酶活性但没有激活 TERT 突变的肿瘤必须通过其他一些机制上调端粒酶。调节端粒酶活性的一种可能方式与端粒位置效应有关,这是一种表观遗传方式,端粒缩短可以通过这种方式增强端粒近端基因(例如 TERT)的表达。需要进一步的工作来确定这是否是在致癌过程中 TERT 转录增加的常见机制。癌细胞是否通过其他机制增加端粒酶活性还有待观察,例如增加端粒的组装、募集或端粒酶的催化活性。 端粒酶减少会引发组织衰竭疾病与癌症中端粒酶上调形成鲜明对比的是,端粒酶功能的种系减少会引发一系列以端粒非常短为特征的组织衰竭疾病。这些被称为端粒生物学障碍 (TBD) 的疾病,其严重程度差异很大,可以在出生时、儿童期甚至中年时出现。典型的端粒酶功能不全疾病最初被定义为先天性角化不良,这是一种多器官、全身性疾病,其特征是皮肤色素沉着异常、指甲营养不良、骨髓衰竭和特定癌症的易感性,包括鳞状细胞癌和白血病。根据最近的遗传和临床发现,现在了解到端粒酶相关基因的突变还可导致特发性肺纤维化、再生障碍性贫血以及肝纤维化和肝硬化(表 1)。  表 1 |端粒酶和端粒相关基因在组织衰竭疾病中发生突变。在端粒生物学障碍中发生突变的端粒酶和端粒相关基因从最常见到最不常见列出。 CST,CTC1–STN1–TEN1; hTR,人端粒酶 RNA; PARN,聚(A)核糖核酸酶; RTEL1,端粒延长解旋酶 1 的调节因子; TCAB1,端粒酶 Cajal 体蛋白 1; 表 1 |端粒酶和端粒相关基因在组织衰竭疾病中发生突变。在端粒生物学障碍中发生突变的端粒酶和端粒相关基因从最常见到最不常见列出。 CST,CTC1–STN1–TEN1; hTR,人端粒酶 RNA; PARN,聚(A)核糖核酸酶; RTEL1,端粒延长解旋酶 1 的调节因子; TCAB1,端粒酶 Cajal 体蛋白 1;TERC,端粒酶RNA成分; TERT,端粒酶逆转录酶;TIN2,TRF1-相互作用核因子2;TRF1,端粒重复结合因子1。 已在患有 TBD 的个体中鉴定出端粒酶全酶的每个组分的突变,并提供了对端粒酶结构和功能的深入了解。 TERC(编码 hTR)或 TERT 基因的杂合突变在 TBD 中很常见,并导致单倍剂量不足(酶水平降低)。 TERC 中的突变通常会减少端粒酶催化作用或降低 hTR 水平。大多数TERC突变发生在 hTR 的 PK/T 结构域内,它结合 TERT 并包含用于逆转录的模板序列。许多这些突变会损害TERT结合并降低端粒酶功能,但会保留hTR表达。hTR 中央 CR4/5 结构域的突变也通过损害hTR-TERT关联来降低端粒酶催化活性。hTR 的 3' H/ACA 区域中的突变通常会通过减少肌张力蛋白结合而导致hTR丢失。TBD患者 TERT基因的种系突变分布在整个开放阅读框中,这与人类癌症中TERT启动子的体细胞突变形成对比。 TERT中的编码突变通常会导致催化活性降低,尽管许多突变保留了催化功能。这些发现表明,TERT突变可能会破坏端粒酶生物学的其他方面,包括蛋白质稳定性、复杂组装或端粒募集。为支持这一观点,TERT TEN域或 TERT C末端的突变会损害其向端粒的募集。与端粒酶募集对端粒维持的重要性一致,TPP1中发现了罕见的突变,这些突变特异性地破坏了它与 TERT 的相互作用,并损害了端粒酶向端粒的募集。核心 dyskerin 复合物 (dyskerin–NHP2– NOP10) 识别 hTR H/ACA 结构域对于 hTR 稳定性和加工至关重要。编码角化不良的基因发生突变会降低 hTR 水平并导致先天性角化不良的 X 连锁形式,并且已在先天性角化不良的常染色体隐性形式中鉴定出 NOP10 和 NHP2 中的罕见突变。在编码肌张力障碍相关辅因子 NAF1 的基因中也发现了罕见的突变,这会导致 hTR 水平降低。在 TBD 中降低 hTR 水平的一种常见方法是通过使 PARN失活的突变。这种突变会导致常染色体隐性遗传形式的特发性肺纤维化、再生障碍性贫血和先天性角化不良,并且通常发生在 PARN的核酸酶或 RNA 结合域中。 PARN 突变会减慢 hTR 的成熟速度,导致 hTR 前体的相对积累和成熟 hTR水平的整体降低。 TBDs中还发现了编码 TCAB1 的基因中罕见的常染色体隐性突变。这些突变位于WD40 重复结构域并干扰 TRiC伴侣复合物对 TCAB1蛋白的折叠,从而导致 TCAB1蛋白丢失。除了这些直接损害编码端粒酶全酶组分的基因突变的突变外,在TIN2、CTC1 和端粒延长解旋酶1(RTEL1) 的调节因子中也发现了 TBD 个体的突变,RTEL1是一种参与端粒延长解旋酶的DNA解旋酶维护端粒完整性。令人惊讶的是,大多数 TBD 引起的突变通过降低 hTR 的水平或功能来影响 hTR,从而强调hTR在端粒酶功能中的核心作用。 结论和未来展望端粒酶进化为在单细胞真核生物中复制染色体末端提供了一种简单的解决方案,但端粒酶复合体在长寿、复杂的后生动物(例如人类)中具有更大的重要性。支持强大的细胞分裂以促进组织更新,同时降低患癌症的可能性,这一根本挑战解释了端粒酶在人类中重要性的提高。在抑制癌症的同时促进细胞更新的要求,结合 RNP 的特殊特性,解释了端粒酶复合物调控的复杂性。这些特征解释了 TERT表达的精细转录控制、精确加工和稳定端粒酶 RNA 的需要、活性复合物组装的调节以及酶向端粒募集的调节。在生物体和组织中,某些细胞被许可表达端粒酶,端粒酶的量随着细胞的分化状态而变化。多层次的端粒酶控制是必不可少的,因为端粒酶活性的增加或减少超出一个狭窄的范围与人类疾病有关。尽管取得了重要进展,但我们仍然缺乏对在正常祖细胞中激活 TERT 转录以及在分化过程中沉默TERT的顺式调节元件和反式作用因子的了解。端粒酶RNA曾被认为是不受管制的,并且以普遍存在的方式表达。TBD的研究揭示了hTR令人惊讶的新调节水平,涉及几种酶在处理其 3' 末端的功能。这些酶控制 hTR 成熟的速度,进而决定其稳态水平。 hTR的成熟由结合 hTR H/ACA 结构域的核心肌张力蛋白复合物决定。因此,后生动物端粒酶 RNA 中 H/ACA 结构域的进化决定了 RNA 加工的机制,同时调节水平并保护 hTR 的 3' 端。基于这些观察,肌张力蛋白复合物可以被认为是端粒酶 RNA 末端加工和稳定复合物。这一特征在人端粒酶的低温电子显微镜结构中视觉上很明显,显示核心肌张力蛋白复合物远离TERT的催化位点。 hTR加工酶PAPD5和PARN可以调节hTR的水平,这代表了TBD患者的潜在治疗途径。调节端粒酶活性的其他潜在方法包括端粒酶组装、端粒募集或使用催化激活剂和抑制剂。 端粒酶上调几乎普遍存在于多种癌症类型中,但现在在 TERT 启动子中具有激活突变的癌症中得到了最好的理解。这些简单的非编码突变为转录因子 GABP 创建了一个新的共有结合位点,因此靶向 GABP 已被建议作为抑制该癌症亚群中 TERT 的一种手段。然而,大多数人类癌症缺乏 TERT 启动子突变,因此对癌症中 TERT 转录的控制仍然知之甚少。这一领域的进展可以提高我们对癌症中 TERT 调节的理解,并为癌症治疗提供新途径。 PDF原文见附件 |
【本文地址】
今日新闻 |
推荐新闻 |